Trial 1: An ongoing international, multi-center, single-arm, single-dose, Phase 1/2/3, open-label, pivotal study of one-time CASGEVY infusion1,2
Key inclusion criteria1,2
- Aged 12 to 35 years
- SCD with ≥2 severe VOCs/year in the 2 years prior to screening
- Eligible for autologous hematopoietic stem-cell transplantation (HSCT)
- Patients aged 12 to 16 years were required to have normal transcranial Doppler (TCD)
Key exclusion criteria1,2
- Available 10/10 human leukocyte antigen-matched related donor
- Prior HSCT
- Clinically significant and active bacterial, viral, fungal, or parasitic infection
- Advanced liver disease, history of untreated Moyamoya disease, or presence of Moyamoya disease that in the opinion of the investigator put the patient at risk of bleeding
- Patients aged 12 to 18 years with any history of abnormal TCD in the middle cerebral artery and the internal carotid artery
- More than 10 unplanned hospitalizations or emergency department visits related to chronic pain rather than SCD-related acute pain crises in the year before screening
44 patients received treatment with CASGEVY in the clinical trial1*
44 patients received treatment with CASGEVY in the clinical trial1*
The data presented here are based on an interim analysis as of June 2023. The safety data are for the full analysis set, defined as the 44 patients who received CASGEVY. The efficacy data are for the primary efficacy set (n=31). This set was defined as all patients who had been followed for at least 16 months after CASGEVY infusion. Patients who had less than 16 months of follow-up due to death or discontinuation due to adverse events related to CASGEVY, or who continuously received RBC transfusions for more than 10 months after CASGEVY, were also included in this set. An additional patient who had less than 16 months of follow-up, but was otherwise determined to be a non-responder for the primary efficacy endpoint, was also included in PES.1
*At the time of the interim analysis, a total of 63 patients enrolled in the trial, of which 58 (92%) patients started mobilization. A total of 44 (76%) patients received CASGEVY infusion.1
†All patients were administered an antihistamine and an antipyretic prior to CASGEVY infusion.1
RBC=red blood cell; SCD=sickle cell disease; VOC=vaso-occlusive crisis.
Patients in the trial had a history of ≥2 severe VOCs/year in the 2 years prior to screening1
In this trial, severe VOCs were defined as an occurrence of at least 1 of the following1:
- Acute pain event requiring a visit to a medical facility and administration of pain medications (opioids or intravenous non-steroidal anti-inflammatory drugs [NSAIDs]) or RBC transfusions
- Acute chest syndrome
- Priapism lasting >2 hours and requiring a visit to a medical facility
- Splenic sequestration
‡Interim analysis conducted based on June 2023 data cut-off date.1
§The PES is a subset of the FAS. The PES was defined as all patients who had been followed for at least 16 months after CASGEVY infusion. Patients who had less than 16 months of follow‑up due to death or discontinuation due to CASGEVY‑related adverse events or who continuously received RBC transfusions for more than 10 months after CASGEVY were also included in this set. An additional patient who had less than 16 months of follow-up, but was otherwise determined to be a non-responder for the primary efficacy endpoint, was also included in the PES.1
The baseline characteristics and demographics are consistent between the PES and the FAS.1
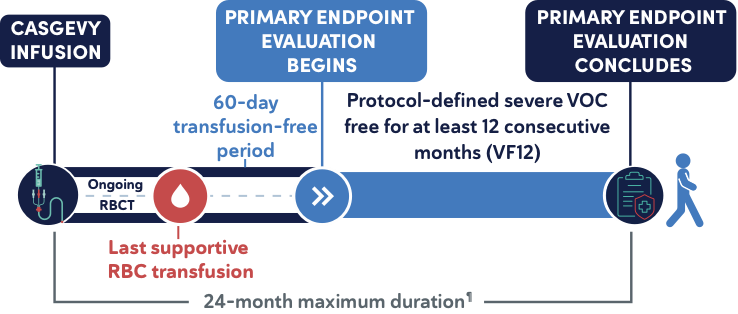
Primary efficacy endpoint1
- Proportion of patients who did not experience any protocol-defined severe VOCs for at least 12 consecutive months (VF12) within the first 24 months after CASGEVY infusion||

Key secondary efficacy endpoint1
- Proportion of patients free from inpatient hospitalization for severe VOCs sustained for at least 12 consecutive months (HF12) after CASGEVY infusion||
Select safety endpoints3
- Successful neutrophil engraftment within 42 days after CASGEVY infusion (Study Day 43)
- Time to neutrophil engraftment
- Time to platelet engraftment
- Safety and tolerability assessments based on adverse events (AEs), clinical laboratory values, and vital signs
Select secondary efficacy endpoints3
- Duration of protocol-defined severe-VOC-free time in patients achieving VF12
- Change in Hb concentrations over time
- Change in HbF concentration over time
- Proportion of alleles with intended genetic modification present in CD34+ cells of the bone marrow over time
- Proportion of alleles with intended genetic modification present in peripheral blood leukocytes over time
||The evaluation starts 60 days after last RBC transfusion for posttransplant support or SCD management. The median (min, max) time to last RBC transfusion after CASGEVY infusion for patients in the primary efficacy set was 19 (11, 52) days.1
¶Postinfusion follow-up includes supportive care, 60-day transfusion-free period after the last supportive RBC transfusion, and the 12 months of the severe-VOC-free period. Patient must have completed this entire postinfusion follow-up within the 24-month study period in order to meet the primary endpoint.1
Hb=hemoglobin; HbF=fetal hemoglobin; RBCT=red blood cell transfusion.
Trial 3: A long-term observational study evaluating the safety and efficacy of CASGEVY for up to 15 years post infusion1,4
Eligibility criteria4
- Patients who received CASGEVY infusion
- Completion of informed consent form and, where applicable, an assent form
Exclusion criteria4
- There are no exclusion criteria
Long-term follow-up primary outcomes4
- New malignancies
- New or worsening hematologic disorders
- All-cause mortality
- Serious AEs occurring up to 15 years post infusion
- AEs related to CASGEVY
Secondary outcomes include long-term efficacy outcomes and patient-reported outcomes.4
Data from this trial are not included in the full Prescribing Information.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Neutrophil Engraftment Failure
Monitor absolute neutrophil counts (ANC) and manage infections according to standard guidelines and medical judgement. In the event of neutrophil engraftment failure, patients should be infused with rescue CD34+ cells.
Delayed Platelet Engraftment
Delayed platelet engraftment has been observed with CASGEVY treatment. There is an increased risk of bleeding until platelet engraftment is achieved. In the clinical trials, there was no association observed between incidence of bleeding events and time to platelet engraftment.
Monitor patients for bleeding according to standard guidelines and medical judgement. Conduct frequent platelet counts until platelet engraftment and platelet recovery are achieved. Perform blood cell count determination and other appropriate testing whenever clinical symptoms suggestive of bleeding arise.
Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis can occur due to dimethyl sulfoxide (DMSO) or dextran 40 in the cryopreservative solution. Monitor patients for hypersensitivity reactions during and after infusion.
Off-Target Genome Editing Risk
Although off-target genome editing was not observed in the edited CD34+ cells evaluated from healthy donors and patients, the risk of unintended, off-target editing in an individual’s CD34+ cells cannot be ruled out due to genetic variants. The clinical significance of potential off-target editing is unknown.
ADVERSE REACTIONS
The most common Grade 3 or 4 non-laboratory adverse reactions (occurring in ≥ 25%) were mucositis and febrile neutropenia in patients with SCD and patients with TDT, and decreased appetite in patients with SCD.
All (100%) of the patients with TDT and SCD experienced Grade 3 or 4 neutropenia and thrombocytopenia. Other common Grade 3 or 4 laboratory abnormalities (≥ 50%) include leukopenia, anemia, and lymphopenia.
DRUG INTERACTIONS
No formal drug interaction studies have been performed. CASGEVY is not expected to interact with the hepatic cytochrome P450 family of enzymes or drug transporters.
Use of Granulocyte-Colony Stimulating Factor (G-CSF): G-CSF must not be used for CD34+ HSC mobilization of patients with SCD.
Use of Hydroxyurea: Discontinue the use of hydroxyurea at least 8 weeks prior to start of each mobilization cycle and conditioning. There is no experience of the use of hydroxyurea after CASGEVY infusion.
Use of Voxelotor and Crizanlizumab: Discontinue the use of voxelotor and crizanlizumab at least 8 weeks prior to start of mobilization and conditioning, as their interaction potential with mobilization and myeloablative conditioning agents is not known.
Use of Iron Chelators: Discontinue the use of iron chelators at least 7 days prior to initiation of myeloablative conditioning, due to potential interaction with the conditioning agent. Some iron chelators are myelosuppressive. If iron chelation is required, avoid the use of non-myelosuppressive iron chelators for at least 3 months and use of myelosuppressive iron chelators for at least 6 months after CASGEVY infusion. Phlebotomy can be used instead of iron chelation, when appropriate.
USE IN SPECIFIC POPULATIONS
Pregnancy/Lactation: CASGEVY must not be administered during pregnancy and breastfeeding should be discontinued during conditioning because of the risks associated with myeloablative conditioning. Pregnancy and breastfeeding after CASGEVY infusion should be discussed with the treating physician.
Females and Males of Reproductive Potential: A negative serum pregnancy test must be confirmed prior to the start of each mobilization cycle and reconfirmed prior to myeloablative conditioning.
Women of childbearing potential and men capable of fathering a child should use effective methods of contraception from start of mobilization through at least 6 months after administration of CASGEVY. Advise patients of the risks associated with conditioning agents.
Infertility has been observed with myeloablative conditioning therefore, advise patients of fertility preservation options before treatment, if appropriate.
Please see full Prescribing Information for CASGEVY.