BCL11A helps mediate the shift from HbF to HbA, repressing γ-globin expression and HbF production3
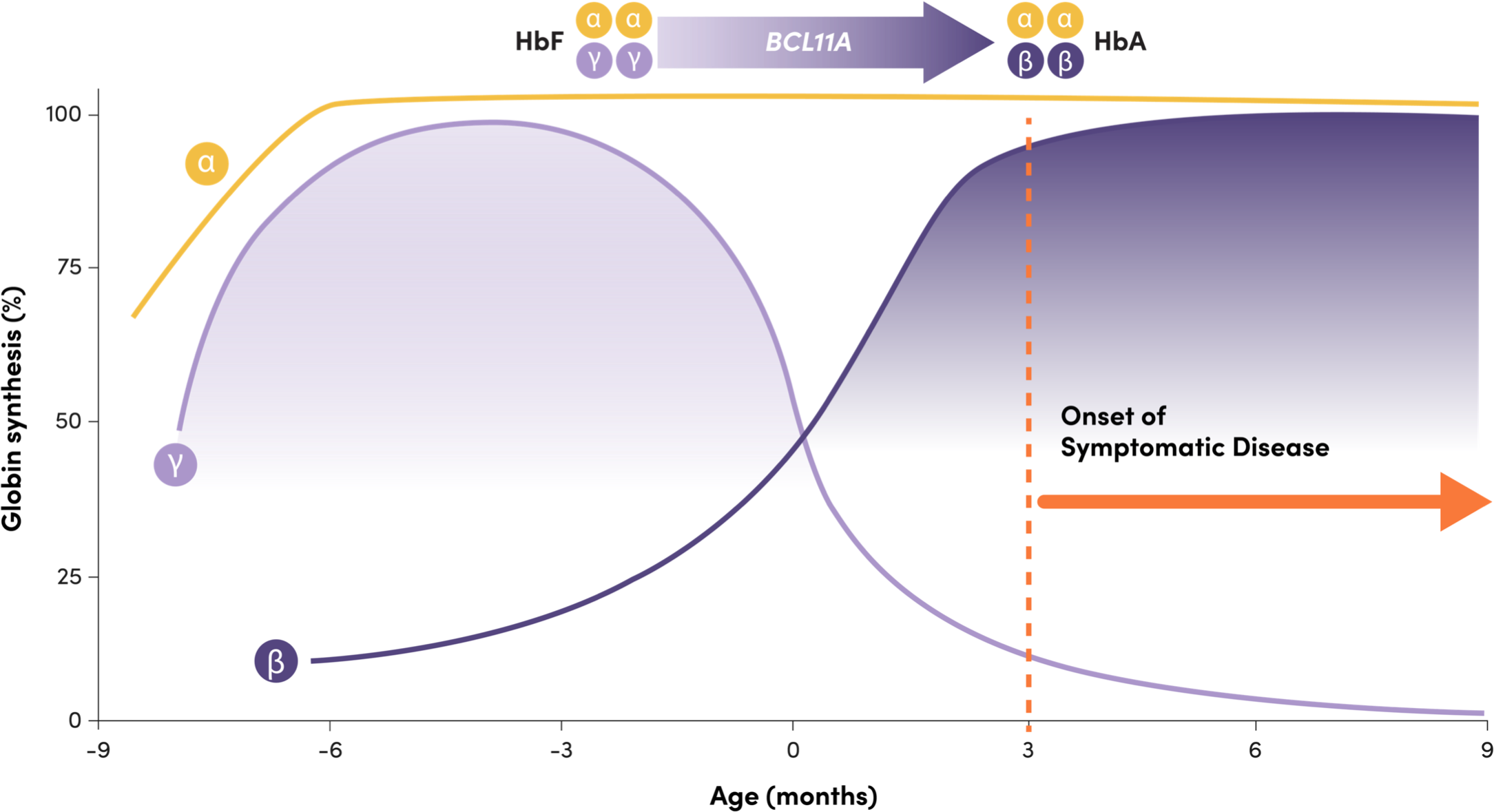
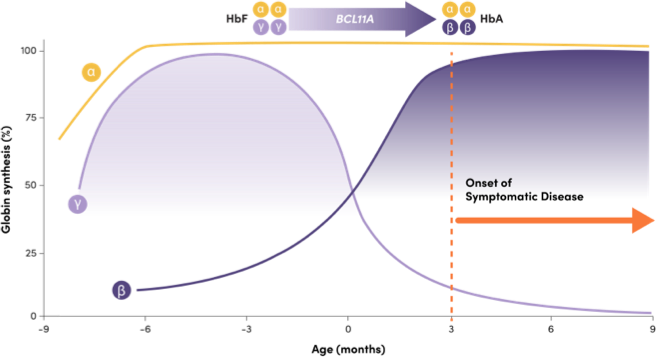
Adapted from Frangoul H, et al. N Engl J Med. 2021;384(3):252-260.
Onset of symptomatic disease
- HbF: 2 γ-globin and 2 α-globin chains, predominant before and at birth1
- HbA: 2 β-globin and 2 α-globin chains, predominant a few months after birth1
- Infants with SCD are typically asymptomatic while their HbF levels remain high, and symptoms arise when HbF declines1,2
- BCL11A is a gene that plays a critical role in the shift, repressing γ-globin expression and HbF production1,4
- Downregulation of BCL11A expression in erythroid cells has been shown to increase HbF production5
- Naturally occurring common genetic variants in the erythroid-specific enhancer region of BCL11A have been associated with increased HbF levels6-8
Natural history studies indicate that elevated levels of HbF are associated with improved health outcomes9,10
Patients with coinheritance of SCD and hereditary persistence of fetal hemoglobin (HPFH) continue to express high concentrations of HbF into adulthood and experience little to no symptoms.1,9,11
HbF levels
| ~30% | Typical in people with coinheritance of SCD and HPFH who are largely asymptomatic1,12 |
| ~8% | Typical in patients with SCD* who may experience VOCs13,14 |
In addition to elevated HbF levels, patients with coinheritance of SCD and HPFH have12,15:
- Pancellular distribution of HbF
- Sufficient HbF to prevent HbS polymerization within each circulating erythrocyte expressing fetal hemoglobin (F cell)

IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Neutrophil Engraftment Failure
Monitor absolute neutrophil counts (ANC) and manage infections according to standard guidelines and medical judgement. In the event of neutrophil engraftment failure, patients should be infused with rescue CD34+ cells.
Delayed Platelet Engraftment
Delayed platelet engraftment has been observed with CASGEVY treatment. There is an increased risk of bleeding until platelet engraftment is achieved. In the clinical trials, there was no association observed between incidence of bleeding events and time to platelet engraftment.
Monitor patients for bleeding according to standard guidelines and medical judgement. Conduct frequent platelet counts until platelet engraftment and platelet recovery are achieved. Perform blood cell count determination and other appropriate testing whenever clinical symptoms suggestive of bleeding arise.
Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis can occur due to dimethyl sulfoxide (DMSO) or dextran 40 in the cryopreservative solution. Monitor patients for hypersensitivity reactions during and after infusion.
Off-Target Genome Editing Risk
Although off-target genome editing was not observed in the edited CD34+ cells evaluated from healthy donors and patients, the risk of unintended, off-target editing in an individual’s CD34+ cells cannot be ruled out due to genetic variants. The clinical significance of potential off-target editing is unknown.
ADVERSE REACTIONS
The most common Grade 3 or 4 non-laboratory adverse reactions (occurring in ≥ 25%) were mucositis and febrile neutropenia in patients with SCD and patients with TDT, and decreased appetite in patients with SCD.
All (100%) of the patients with TDT and SCD experienced Grade 3 or 4 neutropenia and thrombocytopenia. Other common Grade 3 or 4 laboratory abnormalities (≥ 50%) include leukopenia, anemia, and lymphopenia.
DRUG INTERACTIONS
No formal drug interaction studies have been performed. CASGEVY is not expected to interact with the hepatic cytochrome P450 family of enzymes or drug transporters.
Use of Granulocyte-Colony Stimulating Factor (G-CSF): G-CSF must not be used for CD34+ HSC mobilization of patients with SCD.
Use of Hydroxyurea: Discontinue the use of hydroxyurea at least 8 weeks prior to start of each mobilization cycle and conditioning. There is no experience of the use of hydroxyurea after CASGEVY infusion.
Use of Voxelotor and Crizanlizumab: Discontinue the use of voxelotor and crizanlizumab at least 8 weeks prior to start of mobilization and conditioning, as their interaction potential with mobilization and myeloablative conditioning agents is not known.
Use of Iron Chelators: Discontinue the use of iron chelators at least 7 days prior to initiation of myeloablative conditioning, due to potential interaction with the conditioning agent. Some iron chelators are myelosuppressive. If iron chelation is required, avoid the use of non-myelosuppressive iron chelators for at least 3 months and use of myelosuppressive iron chelators for at least 6 months after CASGEVY infusion. Phlebotomy can be used instead of iron chelation, when appropriate.
USE IN SPECIFIC POPULATIONS
Pregnancy/Lactation: CASGEVY must not be administered during pregnancy and breastfeeding should be discontinued during conditioning because of the risks associated with myeloablative conditioning. Pregnancy and breastfeeding after CASGEVY infusion should be discussed with the treating physician.
Females and Males of Reproductive Potential: A negative serum pregnancy test must be confirmed prior to the start of each mobilization cycle and reconfirmed prior to myeloablative conditioning.
Women of childbearing potential and men capable of fathering a child should use effective methods of contraception from start of mobilization through at least 6 months after administration of CASGEVY. Advise patients of the risks associated with conditioning agents.
Infertility has been observed with myeloablative conditioning therefore, advise patients of fertility preservation options before treatment, if appropriate.
Please see full Prescribing Information for CASGEVY.
References: 1. Sankaran VG, Orkin SH. The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med. 2013;3(1):a011643. doi:10.1101/cshperspect.a011643 2. McGann PT, Ware RE. Hydroxyurea therapy for sickle cell anemia. Expert Opin Drug Saf. 2015;14(11):1749-1758. doi:10.1517/14740338.2015.1088827 3. Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med. 2021;384(3):252-260. doi:10.1056/NEJMoa2031054 4. Yin J, Xie X, Ye Y, Wang L, Che F. BCL11A: a potential diagnostic biomarker and therapeutic target in human diseases. Biosci Rep. 2019;39(11):BSR20190604. doi:10.1042/BSR20190604 5. Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322(5909):1839-1842. doi:10.1126/science.1165409 6. Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342(6155):253-257. doi:10.1126/science.1242088 7. Sebastiani P, Farrell JJ, Alsultan A, et al. BCL11A enhancer haplotypes and fetal hemoglobin in sickle cell anemia. Blood Cells Mol Dis. 2015;54(3):224-230. doi:10.1016/j.bcmd.2015.01.001 8. Pule GD, Bitoungui VJN, Chemegni BC, Kengne AP, Antonarakis S, Wonkam A. Association between variants at BCL11A erythroid-specific enhancer and fetal hemoglobin levels among sickle cell disease patients in Cameroon: implications for future therapeutic interventions. OMICS. 2015;19(10):627–631. doi:10.1089/omi.2015.0124 9. Lettre G, Bauer DE. Fetal haemoglobin in sickle-cell disease: from genetic epidemiology to new therapeutic strategies. Lancet. 2016;387(10037):2554-2564. doi:10.1016/S0140-6736(15)01341-0 10. Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. Blood. 1994;84(2):643-649. doi:10.1182/blood.V84.2.643.643 11. Ngo DA, Aygun B, Akinsheye I, et al. Fetal haemoglobin levels and haematological characteristics of compound heterozygotes for haemoglobin S and deletional hereditary persistence of fetal haemoglobin. Br J Haematol. 2012;156(2):259-264. doi:10.1111/j.1365-2141.2011.08916.x 12. Steinberg MH, Chui DH, Dover GJ, Sebastiani P, Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123(4):481-485. doi:10.1182/blood-2013-09-528067 13. Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease: rates and risk factors. N Engl J Med. 1991;325(1):11-16. doi:10.1056/NEJM199107043250103 14. Shah N, Bhor M, Xie L, et al. Evaluation of vaso-occlusive crises in United States sickle cell disease patients: a retrospective claims-based study. J Health Econ Outcomes Res. 2019;6(3):106-117. doi:10.36469/966 15. Steinberg MH. Fetal hemoglobin in sickle cell anemia. Blood. 2020;136(21):2392-2400. doi:10.1182/blood.2020007645